Chapter 2 PreProcessing Data
2.1 Learning Objectives
- Understanding Fastq
- Inspecting and interpreting the quality of sequence data with FASTQC
- Cleaning out sequence data with trimmomatic.
2.2 Fastq
Fastq is a typical sequence format generated by HTS machines. It contains four sections, a sequence ID, the sequence, the ID again and a messy looking quality string made up of characters, each of which represents the quality of the base above it. Here’s an example:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+SEQ_ID
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65Each of the weird characters represents a number according to the ASCII look up table, where numbers are linked to characters, so ! means 33 and " means 34. These numbers are generally Phred scores, which encode the likelihood of the base being wrong on a log scale.
We can use this quality information to assess how well our sequencing went. Along with sequence quality information we should also assess the composition of the sequence data. A program called FastQC (Andrews 2017) is useful for this.
2.2.1 Quality score encoding
Different sequencers use slightly different variations on the Phred score, this is usually called the quality encoding. Older Illumina pipelines encoded a score from -5 to 62 using ASCII characters 59 to 126, but nowadays most use Sanger encoding, which encodes a score from 0 to 93 using ASCII 33 to 126.
Because of this discrepancy, it is necessary to sometimes be explicit about the sequence encoding in Galaxy. We do this by setting the data attributes of data files
2.3 FastQC
FastQC presents a range of plots and summary statistics, you need to provide it with Fastq data.
A typical output like that in Figure 2.1 shows the per base sequence quality.
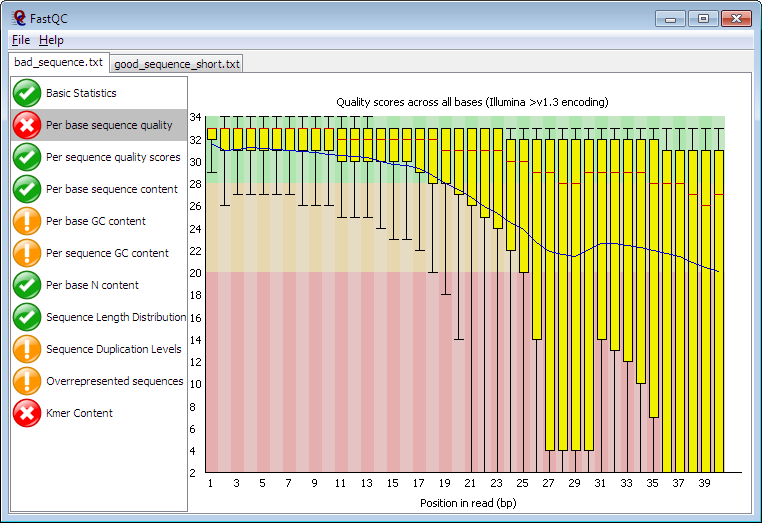
Figure 2.1: FastQC Summary plot. Along the x-axis the plot shows the position in the read and for each position in the reads it shows a box-plot of all the quality scores at that position.
The box plots to the left have much higher and tightly grouped quality scores than those on the right. This is typical of Illumina machine sequence, the quality decreases the further you get along the read. As you can infer from the red region of the plot background, Phred scores less than 20 are generally not trusted.
We may (or may not) decide that we need to get rid of the lower quality sequence.
At the individual sequence read level, we can discard entire sequences if part is too poor or trim the read leaving the good part alone. One system for doing this is (Bolger, Lohse, and Usadel 2014) which can perform a variety of trimming operations on sequence reads. It can remove parts of reads from the left or right sides up to quality thresholds - it uses a sliding window average, rather than just a harsh cut-off.
2.3.1 Sometimes we shouldn’t trim
If we do carry out trimming, then we may end up with lots of reads of different lengths - this can be a problem for some aligners and downstream tools, so sometimes trimming isn’t the best strategy, we have to make context dependent decisions.
At the sequence sample level (e.g, the read file level), we may discover that our read set is not good. Reports from FastQC like the k-mer content plot (Figure 2.2) can show sequence problems, in this graph there is an over-representation of particular k-mers at the start of the sequence that shouldn’t be there.
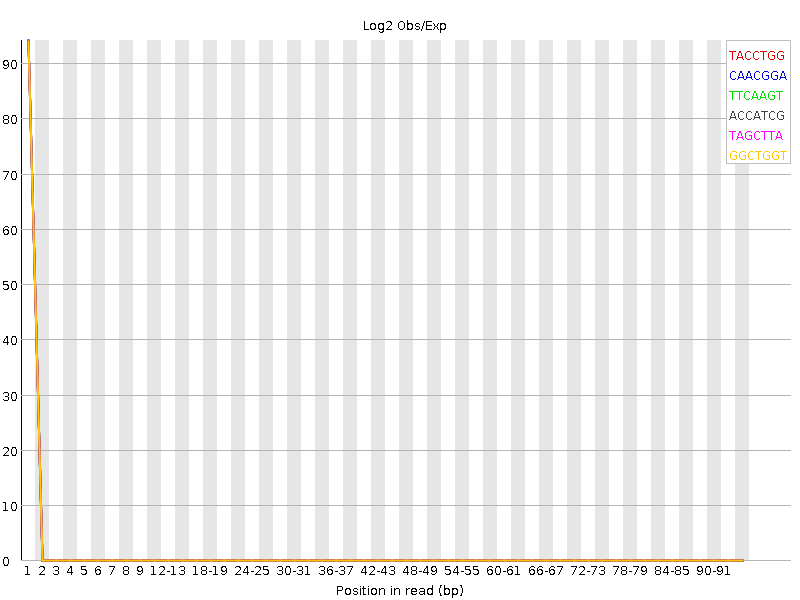
Figure 2.2: The first few bases here are significantly enriched, this can be due to sequence adapters (if they were used) but if not, then the sequence is likely not good, even if the quality scores are fine.
2.4 Exercises
Your task now is to load up Galaxy and run some reads through quality control and trimming, prior to downstream use.
2.4.1 Power Up The VM
- Start
VirtualBoxby double-clicking it’s application icon. - Use
File..Import Applianceand select the??????VM file.
2.4.2 Start Galaxy
Galaxy should appear ready and waiting in the Chromium browser on the desktop in the VM. The bookmarks bar in this browser has all the links you need for this workshop.
2.4.3 Run FastQC
Use the reads in the Pre-processing data library. You will find the FastQC tool in the tool list under HTS QC. The reads are single ended from mutagenised Arabidopsis thaliana. They are Illumina Whole Genome Shotgun reads, the sequence pipeline from our provider should have removed any multiplex adapters and the plants are grown in sterile culture so we aren’t expecting contamination.
Please now complete the section quiz at https://goo.gl/forms/GBnZKO2Yt6hROAvw2.
- How many reads are you using?
- What sort of output files do you get from FastQC?
- What should you do with these files?
- Do they represent a scientific control that could be published?
2.4.4 Interpret Sequence Quality
- Is there any evidence of contamination? Which report tells you?
- If there is, which sequence is contaminating?
2.4.5 Clean Up Poor Quality Sequence
Use the Trimmomatic tool in HTS QC.
- Find and try a trimming strategy to get rid of problems you observed in the section on interpreting sequence quality. Select an appropriate
Average quality required? - Which trimming strategy improves the set of reads?
- How could you filter on size if you needed to pass only good quality, full length sequences to the next step?
References
Andrews, Simon. 2017. FastQC: A Quality Control Tool for High Throughput Sequence Data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Bolger, Anthony M, Marc Lohse, and Bjoern Usadel. 2014. “Trimmomatic: a flexible trimmer for Illumina sequence data.” Bioinformatics (Oxford, England) 30 (15): 2114–20.